
VII. APPROVAL PATH OF NEW PROTOCOLS
When a written protocol is completed to the satisfaction of the PI, AI's, and, perhaps, their Branch Chief or Department Head, it is time to complete form NIH-1195, "Clinical Research Protocol: Initial Review Application," described in chapter I, "Guidelines for Protocol Preparation." The form serves as a conveyance instrument with spaces for all required approval signatures.
The PI forwards the completed NIH-1195 form, the protocol (including consent and assent documents), and all pertinent reference materials to the Accountable Investigator, the Branch Chief or Department Head, and the Clinical Director of the reviewing IRB for their final review, approval, and signature. The Institute Clinical Director may withhold signature until notified of the action taken by the IRB. Pre-IRB scientific review is performed before the initial review package is submitted to the IRB (see below).
Protocol coordinators (PC's) generally work in the office of the Institute Clinical Director or the Chair of the IRB. PC's have copies of all policies and procedures, guides and instructions, and forms and documents, some of which may be specific to their own IRB's. PC's have information on upcoming IRB meeting dates, submission deadlines, number of copies required for submission to the IRB, and protocol review status.
To avoid unnecessary delay in starting a protocol that uses radiation for research, the PI should submit the completed form NIH-88-23(a), "Application for Authorization to Use Radiation in Research Involving Human Subjects," and the protocol, including consent and assent documents, simultaneously to the RSC and the appropriate IRB for concurrent review. See chapter VI, "Protocols Using Radiation for Research," for more detail about RSC review.
Pre-IRB Scientific Review
As directed by the DDIR and required by the NIH MPA, pre-IRB scientific review by the Institute or Center is required for all new protocols. All new protocols are reviewed and considered for the quality, originality, and importance of the question to science or clinical practice; the protocol's relevance to the Institute's mission; and the resources required to support the science. Protocols of a particularly sensitive nature may also warrant review by the DDIR. To signify approval of the initial review package, the designee for the pre-IRB scientific review signs the NIH-1195 form. The PC promptly forwards to the IRB the original initial review package, together with all relevant information and correspondence.
Institutional Review Board Review
45 CFR 46 requires that a committee, at the NIH and elsewhere called an Institutional Review Board, or IRB, prospectively and regularly review and approve human-subjects research activities. The primary mandate of the IRB is to protect and safeguard the rights and welfare of human subjects. To avoid conflict of interest when reviewing a proposal for which its Clinical Director, Scientific Director, or Institute Director is the PI, the IRB may enlist the help of ad hoc reviewers from another Institute or refer the proposal to the IRB of another Institute.
The PC distributes copies of each upcoming initial review package to all IRB members at least a week before the scheduled meeting. Although each IRB has different rules, usually a primary, and perhaps a secondary, reviewer assesses the protocol in detail beforehand. The primary and secondary reviewers report their findings to the IRB at the meeting, at which time the protocol is discussed in detail by all members. PI's are expected to attend the IRB meetings to present new protocols and to answer questions posed by the IRB members. PI's are not necessarily expected to be present for the continuing review of protocols, although the IRB Chair may request their attendance.
The NIH IRB's approve research that satisfies the following minimum requirements:
- Risks to research participants are minimized.
- The risks are reasonable in relation to anticipated benefits to research participants, and to the importance of knowledge that may reasonably be expected to result.
- The selection of research participants is equitable.
- Informed consent will be sought from the research participants or their legally authorized representatives.
Five options are open to the IRB in acting on the initial review package:
- Approval
Approval (without stipulations) of the protocol requires the approval of a majority of the members present at the meeting. If the vote is not unanimous, the minority opinion must be recorded in, or attached to, the minutes.
- Approval with recommendations
The IRB may approve a protocol with nonbinding recommendations. The PI must respond in writing to IRB recommendations, suggestions, and technical corrections. However, the PI may move forward with a protocol that has been approved with recommendations by the IRB.
- Approval with stipulations
The IRB may approve a protocol with specific conditions that must be met by the PI before proceeding with the protocol. The IRB will return the initial review package to the PI, along with written notification identifying its action and specific requests for change. The PI may appeal to the IRB for reconsideration of one or more of the stipulations. However, PI's may not seek to reverse an IRB decision by submitting the same protocol to another NIH IRB. The PI is required to respond, in writing, to IRB stipulations within 30 days. If the stipulations are not addressed within 30 days, the PI will have to resubmit the entire protocol review package for another review. The PI may not move forward with the protocol until the IRB stipulations have been adequately addressed and accepted by the IRB.
Once the stipulations have been addressed, the PI should submit a "Response to IRB Stipulations" memorandum and two copies of the revised initial review package to the PC for review and approval by the IRB Chair. Changes to the protocol or consent and assent documents should be highlighted in one copy of the revised initial review package. Depending on the extent of the stipulations, the IRB Chair may review and approve the revised package without full committee review. The IRB minutes will specify whether stipulations are to be reviewed by the Chair, an IRB subcommittee, or the fully convened IRB.
- Table
The IRB agrees that approval cannot be granted until further information is provided or specific changes are made in the proposed protocol. When the PI submits new information, the protocol is reviewed again by the full IRB.
- Disapproval
The IRB sends a notice to the PI identifying the reasons for disapproval of the protocol. Disapprovals are definitive. The PI may submit a new protocol that reflects due consideration of the reasons for disapproval of the old one.
Final IRB approval of the initial review package is indicated by the signature of the IRB Chair on the NIH-1195 form. The PC promptly forwards the original initial review package, revised protocol, with consent and assent documents, the IRB minutes, and all correspondence to the PCSC for signature by the CC Director, or in the case of off-site protocols, to the OHSR for signature by its Director.
Approval by the Clinical Center Director
Upon receipt of a completed protocol review package, a protocol specialist in the PCSC reviews the package to assure that all required documents are included, all elements and questions have been addressed, and all signatures and initials are present. Common deficiencies noted by the PCSC staff include failure of AI's to initial the NIH-1195 form or protocol face sheet; missing Clinical Director initials for a cross-institutional AI; no IRB minutes; or, in the case of IRB stipulations, a missing formal memorandum titled "PI Response to IRB Stipulations." The protocol specialist works with the Institute PC, via telephone and electronic mail, to resolve any deficiencies. However, it may be necessary to return the protocol review package if it requires major correction.
Once the protocol review package is deemed complete, the protocol specialist abstracts and enters relevant data into the comprehensive computerized data base, called "PROTRAK." PROTRAK contains data on every active protocol involving patients at the CC and is the official source of information about those protocols for all users.
Next, the initial review package is forwarded to the CC Director for review and approval. The Director reviews initial protocols primarily to assess resource use and potential impact on the operations of the CC and its departments. If questions arise during the review, the CC Director may contact the PI directly to discuss and resolve any issues. The Director may also choose to review the issues raised and discussed by the various review bodies during the protocol approval process, as documented in their minutes or correspondence, or the Director may choose to meet with involved parties, such as the IRB Chair, Clinical Director, or Department Head, to discuss key issues. When all issues have been resolved, the CC Director signs the NIH-1195 form, indicating final approval of the protocol. The yearly anniversary date, on which the protocol is due for continuing review, is based upon the CC Director's approval date, found on the NIH-1195 form. This date may be changed only upon recommendation of the IRB Chair and approval by the Director of OHSR. The CC Director returns the approved protocol review package to the PCSC.
Then, the protocol specialist assigns the protocol number, places the protocol consent and assent documents on the intranet, and enters the protocol into the MIS listing of approved protocols. The protocol specialist signs and dates the bottom of the NIH-1195 form, signifying that all tasks are completed and that accrual of subjects can begin. PCSC retains the original NIH-1195 form signed by all the required individuals and review bodies. Copies of the form are sent to the PI, the PC, and to the RSC, if applicable.
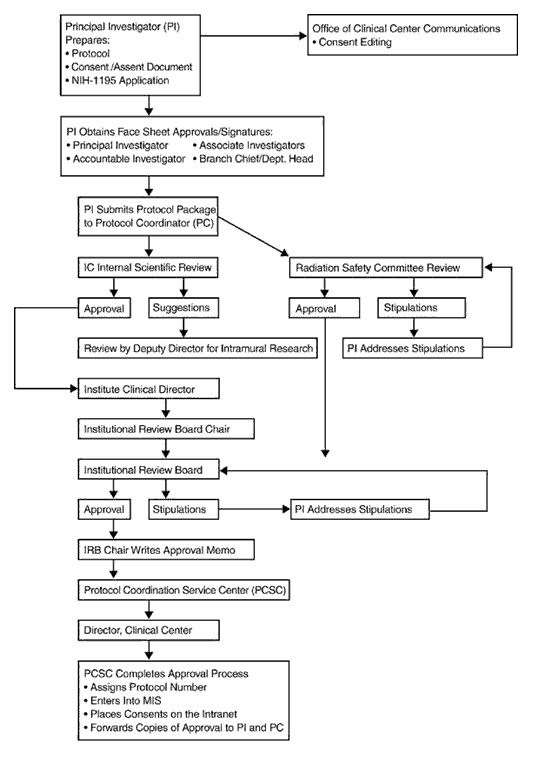
Figure 11 depicts the protocol approval process, from the time the PI prepares the initial protocol through its entry into MIS by PCSC staff.
Figure 11: The Protocol Approval Process
|
|
Expedited Review
The PI can request, in a written memorandum that is attached to the initial review package, that the IRB expedite the review of a protocol of minimal risk. Under expedited procedures, the review is carried out by the IRB Chair or by one or more reviewers designated by the Chair from among the IRB members. The reviewers may exercise all of the authorities of the IRB, except that the reviewers may not disapprove the research without full board review. If approved by expedited review procedures, the usual approval path of new protocols from the Accountable Investigator to the IRB Chair remains unchanged. However, expedited reviews do not require the approval of the CC Director. Nevertheless, the initial review package must still be processed by the PCSC in the same manner as those reviewed by the full IRB. The yearly anniversary date on which the protocol is due for continuing review, having been approved by expedited review, is based on the PCSC completion date. Each instance of expedited review must be reported by the IRB Chair to the full IRB at its next scheduled meeting.
Research activities that present no more than minimial risk to human subjects, and involve only procedures listed in one or more of the categories identified below, may be reviewed by the IRB through the expedited review procedure authorized by 45 CFR 46.110 and 21 CFR 56.110. The activities listed below should not be deemed to be of minimial risk simply because they are included on this list. Inclusion on this list merely means that the activity is eligible for review through the expedited review procedure when the specific circumstances of the proposed research involve no more than minimal risk. The categories apply regardless of the age of subjects, except as noted.
The expedited review procedure may not be used where identification of the subjects and/or their responses would reasonably place them at risk of criminal or civil liability or be damaging to the subjects' financial standing, employability, insurability, reputation, or be stigmatizing, unless reasonable and appropriate protections will be implemented so that risks related to invasion of privacy and breach of confidentiality are no greater than minimal. In addition, it may not be used for classified research involving human subjects. Standard requirements for informed consent (or its waiver, alteration, or exception) apply regardless of the type of review – expedited or convened – utilized by the IRB.
Studies that fall into the following categories are eligible for expedited review, whether initial or continuing:
- Clinical studies of drugs and medical devices only when condition (a) or (b) is met.
- Research on drugs for which an IND application is not required. (Note: Research on marketed drugs that significantly increases the risks or decreases the acceptability of the risks associated with the use of the products is not eligible for expedited review.)
- Research on medical devices for which (i) an investigational device exemption application is not required; or (ii) the medical device is cleared/approved for marketing and the medical device is being used in accordance with its cleared/approved labeling.
- Collection of blood samples by finger stick, heel stick, ear stick, or venipuncture, from the following groups:
- healthy, nonpregnant adults who weigh at least 110 pounds. For those subjects, the amounts drawn may not exceed 550 mL in an 8-week period, and collection may not occur more frequently than 2 times per week; or
- other adults and children, considering their age, weight, and health, the collection procedure, the amount of blood to be collected, and the frequency with which it will be collected. For these subjects, the amount drawn may not exceed the lesser of 50 mL or 3 mL per kg in an 8-week period, and collection may not occur more frequently than 2 times per week.
- Prospective collection of biological specimens for research purposes by noninvasive means.
Examples include: (a) hair and nail clippings in a nondisfiguring manner; (b) deciduous teeth at time of exfoliation or if routine patient care indicates a need for extraction; (c) permanent teeth if routine patient care indicates a need for extraction; (d) excreta and external secretions (including sweat); (e) uncannulated saliva collected either in an unstimulated fashion or stimulated by chewing gumbase or wax or by applying a dilute citric solution to the tongue; (f) placenta removed at delivery; (g) amniotic fluid obtained at the time of rupture of the membrane before or during labor; (h) supra- and subgingival dental plaque and calculus, provided the collection procedure is not more invasive than routine prophylactic scaling of the teeth and the process is accomplished in accordance with accepted prophylactic techniques; (i) mucosal and skin cells collected by buccal scraping or swab, skin swab, or mouth washings; (j) sputum collected after saline mist nebulization.
- Collection of data through noninvasive procedures (not involving general anesthesia or sedation) routinely employed in clinical practice, excluding procedures involving x-rays or microwaves. Where medical devices are employed they must be cleared/approved for marketing. (Studies intended to evaluate the safety and effectiveness of the medical device are not generally eligible for expedited review, including studies of cleared medical devices for new indications.)
Examples include: (a) physical sensors that are applied either to the surface of the body or at a distance and do not involve input of significant amounts of energy into the subject or an invasion of the subject's privacy; (b) weighing or testing sensory acuity; (c) magnetic resonance imaging; (d) electrocardiography, electroencephalography, thermography, detection of naturally occurring radioactivity, electroretinography, ultrasound, diagnostic infrared imaging, Doppler blood flow, and echocardiography; (e) moderate exercise, muscular strength testing, body composition assessment, and flexibility testing where appropriate given the age, weight, and health of the individual.
- Research involving materials (data, documents, records, or specimens) that have been collected or will be collected solely for nonresearch purposes (such as medical treatment or diagnosis).
- Collection of data from voice, video, digital, or image recordings made for research purposes.
- Research on individual or group characteristics or behavior (including, but not limited to, research on perception, cognition, motivation, identity, language, communication, cultural beliefs or practices, and social behavior) or research employing survey, interview, oral history, focus group, program evaluation, human factors evaluation, or quality assurance methodologies.
- Continuing review of research previously approved by the convened IRB as follows:
- where (i) the research is permanently closed to the enrollment of new subjects; (ii) all subjects have completed all research-related interventions; and (iii) the research remains active only for long-term follow-up of subjects; or
- where no subjects have been enrolled and no additional risks have been identified; or
- where the remaining research activities are limited to data analysis.
- Continuing review of research not conducted under an IND application or investigational device exemption, where categories 2 through 8 do not apply, but the IRB has determined and documented at a convened meeting that the research involves no greater than minimal risk, and no additional risks have been identified.
Activities Exempt from Institutional Review Board Review
The MPA and 45 CFR 46 identify six categories of research that, although they involve human subjects, are exempt from the need for IRB review. The OHSR is the only NIH component authorized to determine that a research activity is exempt from the regulations. PI's should seek guidance from OHSR, and not make the determination by themselves, by filling out OHSR's form "Request for Review of Research Activity Involving Human Subjects" at http://ohsr.od.nih.gov/info/Review_Form.php3 (figure 12).
Figure 12: Request for Review of Research Activity Involving Human Subjects
|
|


The six categories of research that do not require IRB review are as follows:
- Research conducted in established or commonly accepted educational settings, involving normal educational practices, such as (a) research on regular and special education instructional strategies, or (b) research on the effectiveness of or the comparison among instructional techniques, curricula, or classroom management methods.
- Research involving the use of educational tests (cognitive, diagnostic, aptitude, achievement), survey procedures, interview procedures, or observation of public behavior, unless: (a) information obtained is recorded in such a manner that human subjects can be identified, directly or through identifiers linked to the subjects; and (b) any disclosure of the human subjects' responses outside the research could reasonably place the subjects at risk of criminal or civil liability or be damaging to the subjects' financial standing, employability, or reputation.
- Research involving the use of educational tests (cognitive, diagnostic, aptitude, achievement), survey procedures, interview procedures, or observation of public behavior that is not exempt under paragraph 2 if (a) the human subjects are elected or appointed public officials or candidates for public office, or (b) Federal statute(s) require(s) without exception that the confidentiality of the personally identifiable information will be maintained throughout the research and thereafter.
- Research involving the collection or study of existing data, documents, records, pathological specimens, or diagnostic specimens, if these sources are publicly available or if the information is recorded by the investigator in such a manner that subjects cannot be identified either directly or through identifiers linked to the subjects.
- Research and demonstration projects that are conducted by or subject to the approval of Department or Agency heads, and that are designed to study, evaluate, or otherwise examine public benefit or service programs, procedures for obtaining benefits or services under those programs, possible changes in or alternatives to those programs or procedures, or possible changes in methods or levels of payment for benefits or services under those programs.
- Taste and food quality evaluation and consumer acceptance studies, (a) if wholesome foods without additives are consumed, or (b) if a food is consumed that contains a food ingredient at or below the level and for a use found to be safe, or agricultural chemical or environmental contaminant at or below the level found to be safe, by the FDA or approved by the Environmental Protection Agency or the Food Safety and Inspection Service of the U.S. Department of Agriculture.
In all these studies, information must be recorded in such a way that no human subject can be identified either directly or through identifiers linked to the individual.
The following research activities also require no human-subjects regulatory oversight and are not subject to the requirements of the MPA:
The collection and study of
- samples from deceased individuals;
- samples collected for diagnostic purposes only;
- samples or data that are available from commercial or public repositories or registries;
- established cell lines that are publicly available to qualified scientific investigators; and
- self-sustaining, cell-free derivative preparations including viral isolates, cloned DNA, or RNA.
Investigators should be aware that even though research with these types of materials is not covered by the MPA, it may be subject to other requirements, such as rules governing technology transfer.
Human Gene Therapy
Protocols involving the use of recombinant DNA in patients require several levels of regulatory review in addition to those required for other investigational agents. When preparing such a protocol, the PI will save time by considering the requirements of all regulatory bodies during the development of preclinical studies and the preparation of the protocol and protocol consent documents. It is useful to review not only the guidelines that regulate all human-subject research, but also the "Guidelines for Research Involving Recombinant DNA Molecules," including updates and amendments, published in the Federal Register. The most current version of these guidelines is available on the ORDA web site at http://www4.od.nih.gov/oba. In the guidelines, "Appendix M. Points to Consider in the Design and Submission of Protocols for the Transfer of Recombinant DNA Molecules into One or More Human Subjects" poses specific questions about DNA clinical research that must be answered by the PI. "Appendix M" applies to research conducted at or sponsored by an institution that receives any NIH support for recombinant-DNA research.
Even though submission of the gene therapy protocol to FDA is the last action required, it is the first agency the PI should contact. This is particularly true if the protocol involves recombinant-DNA research and the PI, not a collaborative company, will be the sponsor of the IND application. FDA welcomes such initial contacts because it increases the likelihood that the PI's final proposal will address most of the FDA's major concerns with the safety aspects of the protocol. Many investigators err in making their first contact with FDA the last item they address after all the other regulatory actions.
While it is not a legal requirement, it is advisable that the PI request, for new gene therapy protocols, a "pre-IND meeting" with the responsible authorities in the FDA Center for Biologics Evaluation and Research (CBER). The working group involved is the Division of Cellular and Gene Therapies. After an initial telephone discussion, the PI should draft a formal letter to FDA requesting a pre-IND meeting on the protocol. The letter must propose an agenda, list who will attend the meeting, identify who will speak at the meeting, and provide the general topic of each presentation. The PI should prepare a concise two- to three-page outline summary of the proposed protocol. It is useful as well to include an early draft of the protocol and the preclinical and/or background clinical studies, but this does not replace the concise summary. All documents must be sent to FDA in triplicate. The FDA notifies the PI of a date for the requested pre-IND meeting, which is generally 1 to 3 months after the formal request. The FDA invites members of its staff who have expertise and regulatory responsibility for any experimental materials, devices, or procedures used in the proposed protocol. The PI should be prepared to discuss and justify all experimental aspects of the protocol with the FDA personnel, since the discussion and regulatory issues will not be limited to the gene-transfer aspects of the proposed protocol. It saves time to be able to incorporate input from the FDA pre-IND meeting or other discussion venues into the final protocol submitted to the IRB.
The initial levels of the regulatory approval process involve two local committees: the PI's IRB and the Institutional Biosafety Committee (IBC). The PI must submit the proposed protocol to the appropriate IRB. The IBC requires IRB approval of the proposed protocol, with or without stipulations, prior to its review. The primary mandate of the IRB is to protect and safeguard the rights and welfare of subjects. The IBC serves to ensure the safety of NIH employees (such as nurses and physicians caring for the patients) and the community at large. The IBC requires detailed biochemical and biological information about the DNA vector to be used in the proposed protocol. Incorporating the information that is requested in "Appendix M. Points to Consider,"described earlier in this section, will encompass most of the issues to be reviewed by the IBC. Both IRB and IBC approvals, with or without stipulations, are required before proceeding.
The next phase of regulatory review involves two national regulatory agencies that need to review and approve the proposed protocol. After receiving IRB and IBC approvals, the PI should submit the proposed protocol simultaneously to FDA and to ORDA. As for any other investigational agent, a formal "Investigational New Drug Application" must be filed that covers the entire process of in vivo or ex vivo genetic modification. Forms FDA 1571, "Investigational New Drug Application [IND]," and FDA 1572, "Statement of Investigator" (appendices E and F, respectively), should be submitted in triplicate to FDA, along with the protocol and supporting materials. Both FDA and ORDA also require a point-by-point written response to "Appendix M. Points to Consider." The required format for the submission, including the specific "Points to Consider" that must be addressed in the documentation, changes frequently; before formulating a submission, the PI should check the current requirements via CBER's website or information lines. ORDA will ask a subset of the members of the RAC to provide a preliminary review of the proposed protocol to determine if the protocol should be subjected to a full review at a full public meeting of the RAC.
The authority for approval of all protocols involving recombinant DNA was relinquished by the RAC to the FDA in October 1997. One option under these regulations is that after the preliminary review of the proposed protocol, ORDA will recommend that the protocol be subjected to FDA review only. The FDA then makes the final determination about whether still to initiate a review of the protocol at a full RAC meeting. Thus, full review by the entire RAC at one of its scheduled quarterly meetings may be requested by either a RAC member or by FDA. If both ORDA and FDA determine that full review by the RAC is not required, then the protocol may be initiated after approval by FDA.
As indicated earlier, the proposed protocol goes to the RAC via ORDA. The ORDA needs to have the proposal, backed by appendices if appropriate, 60 days before the next quarterly meeting of the RAC (if a full review is required). Primary and secondary reviewers are appointed to assess the protocol in detail. The reviewers may pose questions or offer comments on the proposed protocol to which the PI must respond in writing. If it is recommended that the protocol be reviewed by the entire RAC at its quarterly public meeting, the PI must attend that meeting. After its review, the RAC responds with approval, stipulations, requests, or disapproval, much as does the IRB. The NIH Director provides the final approval if a full RAC review is required for the proposed protocol.
Regardless of the review process, the PI must provide annual reports about the protocol to FDA, ORDA, and the IRB.
Contact Form
- About Medtran
- Quality control
- Contact us
- Jobs
- Terminology
- Site map
- Confidentiality
- Personal data protection
- Disclaimer
